1.1 能带简介
在形成分子时,原子轨道构成具有分立能级的分子轨道。晶体是由大量的原子有序堆积而成的。由原子轨道所构成的分子轨道的数量非常之大,以至于可以将所形成的分子轨道的能级看成是准连续的,即形成了能带。晶体中电子所能具有的能量范围,在物理学中往往形象化地用一条条水平横线表示电子的各个能量值。能量愈大,线的位置愈高,一定能量范围内的许多能级(彼此相隔很近)形成一条带,称为能带。
在固体物理学中,固体的能带结构(又称电子能带结构,如图一所示)描述了禁止或允许电子所带有的能量,这是周期性晶格中的量子动力学电子波衍射引起的。材料的能带结构决定了多种特性,特别是它的电子学和光学性质。

1.2能带结构的计算
能带结构目前是采用第一性原理(ab initio)计算所得到的常用信息。大致可以分为价带、禁带和导带三部分(如图一所示),导带和价带之间的空隙称为能隙,用Eg表示。计算材料的能带结构即色散曲线E(k),可以使用Materials Studio或VASP软件进行。以下将介绍用Materials Studio进行能带结构计算的基本步骤,以ZnS半导体为例:
(1)打开Materials Studio界面,点击File→ Import→ Structures→ semiconductors,选择ZnS.msi,得到ZnS.xsd文件,如图二(a)所示;

图二 ZnS晶体结构
(2)变换风格:单击右键→ Display Style→ Atom选项中选择Ball and stick。如图二(b)所示;
(3)结构优化:计算能带结构前需进行结构优化。单击CASTEP Calculation,设置如下图三所示,点击Run进行。优化成功会自动生成GeomOpt文件夹。

图三 结构优化
(4)能带结构计算:点击GeomOpt文件夹中的ZnS.xsd。单击CASTEP Calculation,设置如下图四所示,点击Run进行。得到CASTEP Energy文件夹。

图四 能带结构计算
(5)能带结构图的绘制:点击CASTEP Energy文件夹中的ZnS.xsd→ CASTEP Analysis→ Band structure→ View。得到ZnS Band Structure.xcd,具体能带信息见BandStr.castep,如图五。
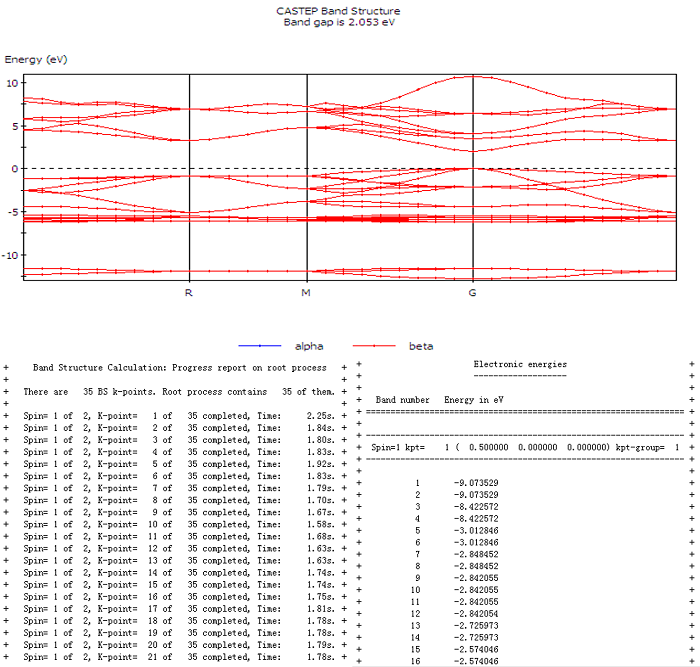
图五 能带结构图
BandStr.castep中记录的信息十分详尽,包括电子数目(自旋向上与自旋向下)、能带数目、计算耗时等。
(6)初步分析:
从能带结构图中得到的信息,判断直接带隙或间接带隙、带隙、价带顶与导带底能量。在origin操作的具体步骤是:把能带图拷贝到Origin中→ 全选→ 作图,得到图六(a)。

图六 能带结构图
具体分析如下:
①导带底与价带顶都在k空间的Γ点上,所以ZnS晶体为直接带隙。
②价带顶位于0 eV,导带顶位于2.053 eV。
③带隙为2.053 eV。
④由CASTEP Calculation中可看出布里渊区中k空间的路径由X→R→M→G,如图六(b)。
【态密度图的绘制及初步分析】
2.1 态密度简介
原则上讲,态密度可以作为能带结构的一个可视化结果。很多分析和能带的分析结果可以一一对应,很多术语也和能带分析相通。但是因为它更直观,因此在结果讨论中用得比能带分析更广泛一些。在电子能级为准连续分布的情况下,单位能量间隔内的电子态数目。即能量介于E~E+△E之间的量子态数目△Z与能量差△E之比,即为态密度。能态密度与能带结构密切相关,是一个重要的基本函数。固体的许多特性,如电子比热、光和X射线的吸收和发射等,都与能态密度有关。
2.2 态密度(DOS)的计算
态密度图可以反映出电子在各个轨道的分布状况,反映原子与原子之间的相互作用情况并且还可以揭示化学键的重要信息。态密度有分波态密度(PDOS)和总态密度(TDOS)两种形式。以下将介绍用Materials Studio进行态密度计算的基本步骤,仍以ZnS半导体为例:
(1)前三步与计算能带结构的一致。
(2)点击GeomOpt文件夹中的ZnS.xsd。单击CASTEP Calculation,设置如下图七所示,点击Run。得到ZnS CASTEP Energy(2)文件,其中包含计算的结果。

图七 DOS与PDOS的计算
(3)打开其中的ZnS.xsd→ CASTEP Analysis→ Density of states → Full DOS→ View。得到TDOS,可以将图形拷贝到Origin中,如下图所示。

图八 DOS的计算
(4)分波态密度的计算:打开其中的ZnS.xsd→ CASTEP Analysis→ Density of states → Partial勾上→ 选择s、p、d、f。得到ZnS PDOS.xsd。可以将图形拷贝到Origin中,如下图十所示。(注意:因为不是在origin的workbook表格里不能都选中,这样会有杂线,操作如下,不要选择杂线的列)

图九 PDOS的设置

图十 PDOS的作图
注意你要标注一下各个颜色对应的轨道(s,p,d)。从对比图可以看出总的态密度由各自哪些轨道贡献的。
(5)态密度图的初步分析:将PDOS图与DOS图放在一起对比,如图十一所示。
①DOS图也可分析能隙特性:若Fermi能级处于DOS值为零的区间中,说明该体系是半导体或绝缘体;若有分波DOS跨过费米能级,则该体系是金属。而两个尖峰之间的DOS并不为零。赝能隙直接反映了该体系成键的共价性的强弱:越宽,说明共价性越强。
由图中可知,Fermi能级处于DOS接近0但又不全为0。说明,该晶体大部分显半导体性质,但金属性较强。
③观察DOS由各自哪些轨道贡献的:Fermi能级左侧为价带,主要由d、p轨道组成,s轨道贡献也有一部分;Fermi能级右侧为导带,主要由s、p轨道组成。
如果具体要分析是Zn还是S的那个轨道对DOS贡献较大仍需进行下一步。

图十一 DOS与PDOS的初步分析
(6)每个原子的PDOS:操作步骤如下,打开ZnS.xsd→ 选定Zn(或S)原子→ 进入CASTEP Analysis → 进行如下设置→ View。依次得到Zn与S的PDOS图,如下。

图十二 单个原子的PDOS分析

图十三 分别是Zn和S的PDOS图
分析:将以上两个PDOS与总的DOS进行对比
①价带:Zn的d轨道与S的p轨道是价带的主要组成来源,S的s轨道有小部分贡献。
②导带:Zn的s、p轨道与S的p轨道是导带的主要来源。
【总结】
能带结构与态密度的分析都是用于探究固体的结构性质的手段,它能很好的预测材料的性质(如成键的趋势、化学键的组成等)、用理论去解释实验现象。

